Research
Identification and characterization of viral
cis-regulatory elements
To withstand a repertoire of antiviral defenses and replicate, viruses must sense, interpret, and respond to many internal and external cues. Retroviruses and DNA viruses can intercept these cues impinging on host transcription factors via cis-regulatory elements (CREs) in viral genomes, allowing them to sense and coordinate context-specific responses to varied signals. Our lab has identified ~3,000 viral CREs across 27 dsDNA viruses from the Adenovirus, Herpesvirus, Polyomavirus, andPapillomavirus families and 5 retroviruses. Using a combination of experimental and in silico approaches we are beginning to determine the mechanism of regulation of these CREs and the cellular states to which these elements respond. Altogether, this work aims to address key questions in viral pathogenesis and disease treatment: 1) Which are the functional differences between human and viral CREs? 2) To which signals do each CREs responds and how is this related to viral reactivation or pathogenesis? 3) What is the extent of functional variation in CREs across isolates and how is this associated with viral plasticity and pathogenicity? 4) How can we leverage viral CRE responses to prevent reactivation or for precision sensitizing therapeutics?


Viral transcriptional regulators
Viral genomes encode transcriptional regulators that control the expression of viral genes and participate in viral reactivation cascades. In addition, these viral transcriptional regulators (vTRs) can be recruited to the host genome and rewire cellular networks to drive cell states that promote viral replication and immune evasion mechanisms. Our lab aims to determine the mechanisms by which vTRs affect host gene expression by integrating multiple high-throughput approaches to measure protein-DNA interactions, protein-protein interactions, transcriptional activity, and transcriptional perturbations. In addition, we aim to identify small molecules that inhibit vTR activities which could lead to novel antiviral therapeutics.
Structure and Function of Immune Gene Regulatory Networks
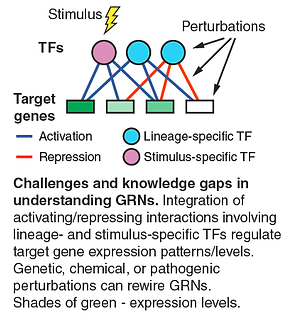
Gene regulatory networks (GRNs) are central to almost all biological processes. Our search focusses on understanding the structure and logic of human GRNs with the ultimate goal of devising strategies for therapeutic interventions. Current gaps in our understanding of GRNs include: determining how combinations of transcription factors (TFs) regulate specific target gene expression patterns, identifying mechanisms by which different genes are co-regulated to effect a given biological response, determining how GRNs are rewired in response to environmental cues, and designing strategies to manipulate GRNs to modulate biological outcomes. Cytokines present an ideal model system to study GRNs because cytokines genes are highly regulated at the transcriptional level and because this regulation involves a complex interplay between cell type-specific TFs and TFs activated by different signaling pathways. In the lab, we tackle the challenge of studying the structure and regulatory logic of the cytokine GRN by integrating complementary methods to map protein-DNA interactions, functional perturbations, and phenotypic characterizations. Our long-term goal is to answer central questions in immune regulation such as: through which mechanisms are immune genes regulated during inflammatory processes? How do viral pathogens perturb immune GRNs? How can we modulate the GRN to more effectively respond to infectious diseases and pathological conditions? Overall, our work will identify general principles and generate a framework to study and manipulate GRNs which will ultimately lead to novel strategies impacting human health.
Gene regulatory network rewiring by TF isoforms
Genome alterations such as amplification, deletion, translocations and mutations, are often considered primary events of cancer progression. However, cancer-specific isoforms resulting from alternative splicing, alternative transcription start sites, and alternative transcriptional termination sites, have also have functional impact on tumorigenesis. In particular, changes in gene regulatory networks by TF isoforms have been shown to play a major role in tumorigenesis and metastasis in multiple types of cancer. While a few examples of functional characterization of driver cancer-specific TF isoforms have been reported, what remains unclear is the extent to which differences in TF isoforms between normal and cancer tissue affect global gene regulatory networks and how such regulatory network rewiring leads to altered gene expression programs in cancer. In collaboration with the Vidal (DFCI), Bulyk (BWH and Harvard), and Salominis (Cincinatti Children’s Hospital) labs, we are characterizing and modeling the effect of large numbers of cancer-specific TF isoforms in the context of interactome networks. We are integrating network modeling and high-throughput systematic experimental strategies at the level of molecular protein-protein and protein-DNA interactions to predict cancer drivers and suppressors. This project constitutes an important step towards the long-term goal of contextualizing and functionalizing large numbers of TF isoforms implicated in cancer which will ultimately lead to novel therapeutics. options.

Characterization of the gene regulatory network governing the first cell fate decision in mammalian embryonic development
Although the “master regulators” that maintain the self-renewal of pluripotent embryonic stem cells and extraembryonic trophoblast stem cells are relatively well understood, little is known about how these transcription factors are regulated in vivo at early stages of embryogenesis. Molecular genetics and embryology studies have identified signaling pathways required to direct cells to become trophoblast or embryonic epiblast, but the transcription factors that activate expression of pluripotency master regulators (PMRs) or trophoblast master regulators (TMRs) in each cell remain largely unknown. We are performing high-throughput experiments to comprehensively identify transcription factors regulating PMRs and TMRs. In collaboration with the Fazzio (UMMS) and Boskovic (EMBL) labs, we are also generating a lineage-resolved atlas of epigenetic changes that occur as cells select either the epiblast or trophoblast fate, and we are identifying the direct targets of transcription factors that participate in cell fate specification. We aim to dissect the mechanisms by which developmental transcription factors help elicit these epigenetic changes and generate a model of the totipotent GRN that mediates this decision. These studies will reshape our understanding of early embryonic gene regulation, as well as specification of epiblast and trophoblast cell fate.

Functional Characterization of Cancer Noncoding Variants
Cancer driver mutations in noncoding regions are difficult to identify in the context of thousands of passenger (non-functional) mutations. Additionally, in most cases, the mechanisms by which noncoding disease variants affect gene regulation have not been determined. Our goal is to tackle these open problems in Biomedical research by generating an end-to-end pipeline to identify and functionally characterize hundreds of cancer driver noncoding variants. This includes developing computational tools to predict driver variants, validating these variants using massively parallel reporter assays, and determining transcription factor binding differences between alleles by using an enhanced yeast one-hybrid approach we recently developed that overcomes many of the limitations associated with chromatin immunoprecipitation and motif binding predictions. These studies will provide mechanistic insights into how noncoding variants alter gene expression in cancer, and may provide personalized therapeutic options.
